需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)發
來源/國家藥品監督管理局官網
9月18日,國家藥監局發布《國家藥監局關于發布需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)的通告(2020年第61號)》及《需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)》修訂說明。全文如下。
國家藥監局關于發布需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)的通告(2020年第61號)
為貫徹落實中共中央辦公廳、國務院辦公廳《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》和國務院深化“放管服”改革要求,進一步加強醫療器械臨床試驗的管理,維護醫療器械臨床試驗過程中受試者權益,推進監管科學研究成果轉化,提高審批效率,加快產品上市,根據《醫療器械監督管理條例》,國家藥品監督管理局組織對需進行臨床試驗審批的第三類醫療器械目錄進行了修訂,現予發布,自發布之日起施行。
《關于發布需進行臨床試驗審批的第三類醫療器械目錄的通告》(國家食品藥品監督管理總局通告2014年第14號)同時廢止。
特此通告。
附件:需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)
國家藥監局
2020年9月14日
附件
需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)
與境內外已上市產品相比,采用全新設計、材料或機理,和/或適用于全新適用范圍,且對人體具有較高風險的醫療器械,應當經臨床試驗審批后方可在中國開展臨床試驗。
上述原則適用的具體品種類別如下:
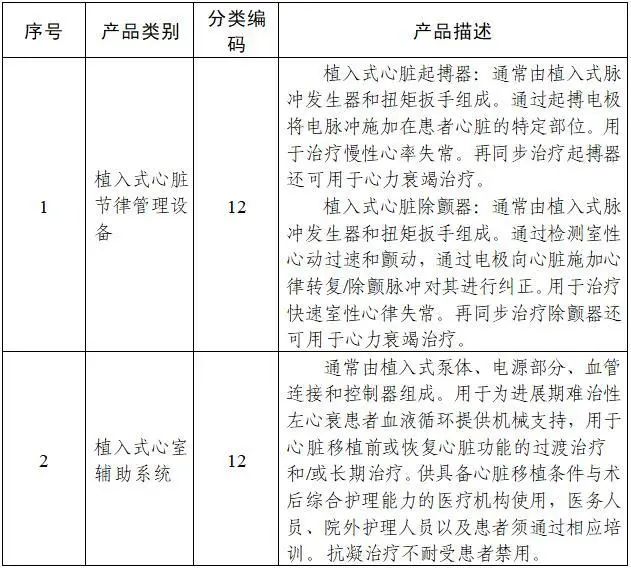

《需進行臨床試驗審批的第三類醫療器械目錄(2020年修訂版)》修訂說明
2014年10月1日,原國家食品藥品監督管理總局組織制定發布了《需進行臨床試驗審批的第三類醫療器械目錄》,對規范醫療器械臨床試驗的開展發揮了積極作用。為貫徹落實中共中央辦公廳、國務院辦公廳《關于深化審評審批制度改革鼓勵藥品醫療器械創新的意見》和國務院深化“放管服”改革要求,進一步加強醫療器械臨床試驗的管理,維護醫療器械臨床試驗過程中受試者權益,推進監管科學研究成果轉化,提高審批效率,加快產品上市,根據《醫療器械監督管理條例》,國家藥品監督管理局組織對需進行臨床試驗審批的第三類醫療器械目錄進行了修訂,發布了《需進行臨床試驗審批的第三類醫療器械目錄》(2020年修訂版)。修訂的目錄增加了適用產品的共性原則描述,調整了6項目錄產品描述,刪除了2項產品,更新了分類編碼。具體包括:
一、增加適用產品的共性原則描述,即“與境內外已上市產品相比,采用全新設計、材料或機理,和/或適用于全新適用范圍,且對人體具有較高風險的醫療器械,應當經臨床試驗審批后方可在中國開展臨床試驗。”調整后的目錄以“共性原則+產品描述”的形式提供,同時滿足兩部分內容要求,則可以判定為本目錄適用產品。
此外,原目錄中部分產品描述為“境內市場尚未出現的”或未明確限定,修訂后目錄將共性原則確定為“與境內外已上市產品相比”,更加符合“風險—受益”評價理念和監管科學要求。
二、調整部分產品類別和描述。將“植入式心臟起搏器、植入式心臟除顫器、植入式心臟再同步復律除顫器”調整為“植入式心臟節律管理設備”,“植入式血泵”調整為“植入式心室輔助系統”,“植入式藥物灌注泵”調整為“植入式藥物輸注設備”,“境內市場上尚未出現的血管內支架系統”調整為“人工心臟瓣膜和血管內支架”;將“境內市場上尚未出現的植入性人工器官、接觸式人工器官、骨科內固定產品及骨科填充材料”調整為“含活細胞的組織工程醫療產品”;將“可吸收四肢長骨內固定產品”調整為“可吸收四肢長骨內固定植入器械”。相應的產品描述也進行了細化和明確。
三、刪除部分產品類別。刪除“定制增材制造(3D打印)骨科植入物”和“納米骨科植入物”。
四、更新醫療器械分類編碼。根據《醫療器械分類目錄》,明確了產品所屬分類目錄中子目錄編碼,考慮到部分產品尚無一級或二級目錄,為了統一分類編碼表述,暫不細化到一級和二級目錄。

